Background:
Spinal muscular atrophy is an autosomal recessive genetic disorder. Type I (Werdnig-Hoffman, acute) is the most severe form of SMA. Onset is noted at 0-3 months due to hypotonia and severe weakness, feeding difficulties and/or need for respiratory support. Type I SMA has historically been characterized as fatal, with lifespan reported as 1 or 2 years.1,2 With current improved care and respiratory support, many children are living much longer. Some children live up to age eight and are able to attend school with appropriate support and accommodations.3
Children with SMA have a reduced number of anterior horn cells in the spinal cord, and there is progressive deterioration of the ones that are present, which leads to progressive loss of strength and function.
The genetic defect in SMA is the survival motor neuron (SMN) gene on chromosome 5q11.2-13. SMN codes for SMN protein, which maintains the anterior horn cells. In the absence of adequate SMN protein, the anterior horn cells die. Diagnosis can be confirmed by EMG, biopsy, or more commonly genetic testing. Upon muscle biopsy, noted are changes characteristic of diseases involving denervation and many atrophic fibers among groups of normal or hypertrophic fibers.1,2
Common impairments and functional limitations in children with Type I SMA1,2,3:
- significant weakness of the neck, spine and extremities
- proximal>distal weakness
* great risk for development of scoliosis
* risk of contracture formation due to persistent gravity-dependent positioning
* respiratory distress and use of compensatory “belly breathing” or paradoxical breathing
* decreased ability or inability to tolerate upright positioning due to oxygen desaturations and inability to manage secretions.
Objectives/Purpose:
Disease modifying treatment with an antisense oligonucleotide (Spinraza) is now available for Spinal Muscular Atrophy (SMA), and gene therapy for SMA is also currently in clinical trials with exciting preliminary results reported. If Spinraza or gene therapy is administered in the newborn period, before cell death has occurred, the potential exists for children with SMA type 1 to sit, stand and/or walk. Previous to this treatment, most children with SMA type 1 passed away in early childhood and never gained the ability to sit. These developments in disease-modifiying treatments are enhancing the potential to gain levels of strength and functional mobility never before possible for patients with SMA, resulting in an imperative to adjust goals and expectations in light of this new treatment.
The ability to work on supported sitting and standing with children with SMA type 1 and 2, has been possible even pre-Spinraza, in the presence of stable vital signs, but may not have been pursued in the absence of an awareness of the potential for more preventative, anticipatory care following a vision of optimizing care in the presence of progressive disorders. Historically, children may have been discharged from therapy for lack of progress or may have only receiving stretching and positioning intervention. For children with SMA type 1, reclined positioning in adaptive strollers or in bed may have been the standard, due to perceived and real difficulties breathing when more upright. Historically, many patients with SMA type 1 passed away before the age of 2, so more aggressive therapies were not commonly utilized. Increased awareness of anticipatory and preventative management, coupled with the exciting evolution of genetically based disease modifying treatments are changing the world for individuals with SMA, The potential for individuals with SMA to gain strength and functional mobility with the emergence of these treatments, coupled with optimal care, has not yet been fully realized, since this treatment is in its infancy, with hopes high and great excitement for the future.
I created, with the assistance of my advisory committee, a poster presentation describing the progress of my patient with SMA type 1 prior to initiation of the disease-modifying treatment (Spinraza). It is intended to educate other pediatric Physical Therapists on their ability to work on more aggressive strengthening and functional mobility with these patients, especially in light of the new disease-modifying therapy available.
Products:
- Poster Presentation
- Application to submit poster presentation to APPTAC
Evaluation:
I received ongoing evaluation from my committee members throughout the process of creating the poster presentation. I will receive feedback from the APPTAC review committee after I submit the poster for acceptance to the conference.
Self-Evaluation:
I am very pleased with and proud of the final project. I am extremely proud of this patient and all he has been able to achieve with the input of therapists, his family and his own motivation to be independent. I now wish that I had published more poster presentations on more patients during my career and hope that this is just the first of many of my contributions to the pediatric physical therapy body of knowledge.
Acknowledgements:
I am indebted to this patient and his family, for always pushing forward and sharing a vision of what is possible rather than what is not possible. The assistance received from my mentors goes far beyond this project and has helped to develop me into the therapist that I am today. I respect my mentors not only for their immense knowledge in their practice areas, but even more importantly for their inspiration, vision, care and compassion towards the patients and families that they encounter. I am beyond lucky to be able to learn from them. Thank you from the bottom of my heart: Laura Case, PT, DPT, MS, PCS, C/NDT, Andrea Hartzell, PT, DPT, MS and Karen McCulloch, PT, PhD, NCS. Thank you also to my supervisors at Duke, past and present for encouraging and supporting me along this journey. I am lucky to call you friends and mentors: Jan Fitch, PT, Robbin Newton, MA, OTR/L, BCP, C/NDT and Andrea Hartzell, PT, DPT, MS.
Finally, thank you to my family for supporting me all along the way. James, Kelly and Jonathan Coats, you inspire me beyond words and I ever so greatly appreciate and love you. Kelly and JJ-I’ll go by Dr. Mom now 🙂
References:
- Campbell SK, Vander Linden DW, & Palisano RJ (2006). Physical therapy for children (3rd ed.). Philadelphia, PA: Saunders Elsevier.
- Farrar MA, Kiernan MC. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. 2015;12(2):290-302.
- Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J. Child Neurol. 2007;22(8):1027-1049.
2 Responses to “Unexpected Outcomes In A Child with SMA type 1 (Pre-Spinraza treatment)”
Julie Coats
Hi Martha,
Thank you for reaching out. Best of luck in your career in pediatrics! Children with SMA are often hospitalized for respiratory issues, so it is likely that you may work with a patient with SMA while inpatient. They also commonly undergo spinal surgery. One of our orthopedists has begun using MAGEC rods, which grow over time with a magnetic external remote control to prevent as significant progression of spinal deformity over time.
I’m so glad that the poster presentation was helpful, please let me know if any more questions arise as you are on your rotation!
As far as activity tolerance, with this patient we have progressed with activity as tolerated. He does not require an oxygen monitor, but I did assess his O2 saturations during initial trials of a SPIO vest. Recently, he has begun to be able to propel a toddler tricycle independently. One day, he propelled for upwards of an hour at home and was visibly fatigued at his therapy session. I let his family know that he may need a break during prolonged activities such as this to avoid excessive fatigue.
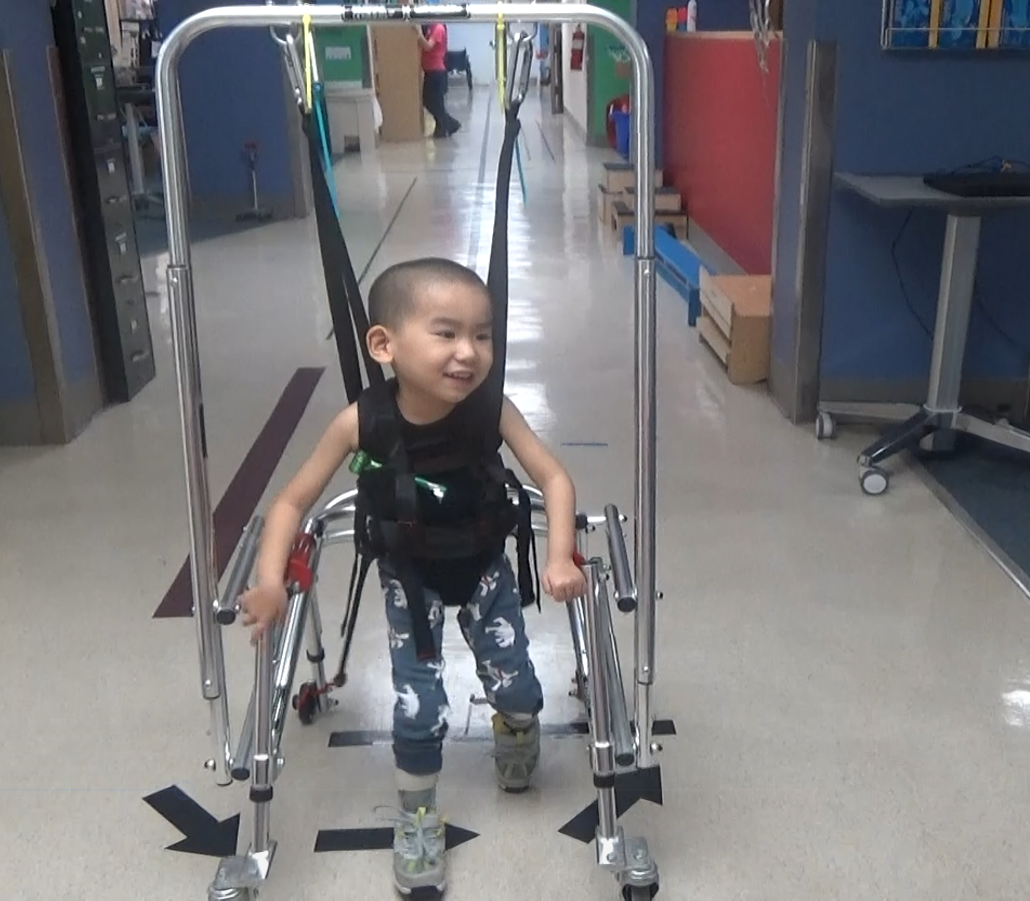
To gain initial head control, we worked on progressive isometric holding over time. We utilized the Head Pod initially to allow appropriate alignment of his head over his body when upright in devices such as the Kaye Body weight support walker. We have worked on progressive strengthening as tolerated, with and without resistance. He has worked on improved postural control in static and dynamic sitting. He has worked on maintaining postures (prone on extended elbows, quadruped, supported standing) and transitioning into and out of these positions. We have emphasized maintenance of musculoskeletal alignment over time.
I hope that some of these suggestions are helpful in your internship and career, thank you for your input!
-Julie
Martha Kalisz
Hi Julie,
I’m definitely impressed with your capstone project! I am interested in pursuing a career in pediatric therapy, and will be completing my final clinical rotation in an acute pediatric setting. I wonder if I’m going to see any children with SMA on my rotation this summer. I don’t know much at all about SMA, but am happy to hear that with disease modifying treatments, children with SMA have been able to live longer. Your project also exposed me to the CHOP-INTEND outcome measure, which I had not previously heard of. Your results with this case seem to indicate the importance of PT in this population as these children are able to live longer – definitely exciting! I’m intrigued by the specific interventions that you used with this child to progress him towards upright activity, and how you measured his activity tolerance. Thanks for your work on this project – great job!